Research Interests
Protein Folding
Over the years, the general theme of this lab has been the study of the mechanisms by which proteins form their three-dimensional structures. In our earlier work, we used a combination of biochemical, biophysical and mutational methods to study the folding of proteins that are stabilized by disulfide bonds, especially bovine pancreatic trypsin inhibitor (BPTI) and an omega-conotoxin. By taking advantage of the ability to physically isolate and characterize the disulfide-bonded intermediates in the folding of these proteins, we were able to study in great detail the effects of amino acid replacements on the folding mechanism. These studies, and the more recent work described below, are directed towards the long term goal of understanding the relationship between a protein's amino acid sequence and it's three-dimensional structure. In addition, these studies have important implications for understanding the effects of mutations on protein structure and function, including mutations that give rise to a variety of human diseases.Unfolded Proteins
Over the past several years, our interests have shifted somewhat, from the process of folding to the properties of unfolded states. In addition to the importance of unfolded proteins as the starting point for folding and the reference state for most measurements of protein stability, non-native states are now known to play important roles in normal cellular processes, including protein localization and degradation, as well as pathological processes such as the formation of amyloid fibers associated with many neurodegenerative diseases. We are using a combination of experimental methods, including small-angle scattering and NMR spectroscopy, and computer simulations to study both the denatured state ensembles of folded proteins and intrinsically disordered proteins (IDPs), a recently-recognized class of of proteins that spend at least part of their time in vivo in a disordered state.
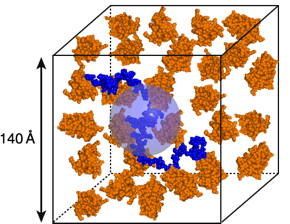
The figure above is a cartoon representation of one of the unfolded proteins we study, the N protein of bacteriophage lambda (blue), in a solution containing about about 130 mg/mL of a small globular protein (orange). The blue sphere has the same volume as the unfolded protein. We are using small-angle neutron scattering (SANS) to study the behavior of unfolded proteins under conditions like those depicted in the figure, and we use computer simulations to test various ideas about unfolded proteins. One of the important results to emerge from these studies is the finding that crowding causes surprisingly little effect on the average dimensions of the N protein, and likely other unfolded proteins.
Protein Dynamics and Protease Inhibitor Function
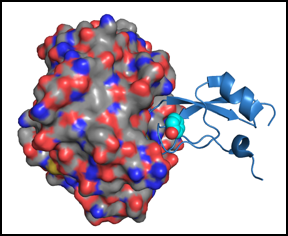
We are also interested in the dynamic properties of folded proteins and how motions, or the lack thereof, are related to function. In particular, we have used NMR spectroscopy to study the effects of mutations on the dynamics of BPTI in its folded and partially folded states. These studies have revealed that specific amino acid residues play particularly important roles in maintaining the relatively fixed conformation of the native state, particularly in the region of the protein that binds to and inhibits serine proteases. Some of the mutations that increase the flexibility of the trypsin binding region also increase it's susceptibility to cleavage by the enzyme.In collaboration with Martin Horvath, we have also used X-ray crystallography to study the structures of complexes between mutant inhibitors and trypsin. The figure below shows the crystal structure of a BPTI variant (the blue ribbon diagram) bound to trypsin (shown as a surface representation). In this particular mutant, Tyr35 of BPTI is replaced with Gly. Solution NMR studies of the free inhibitor have shown that this substitution causes increased flexibility of the chain segments directly involved in trypsin binding. Once bound to the inhibitor, however, the inhibitor becomes nearly as rigid as the wild-type protein, and a cavity is formed in the space normally occupied by the Tyr side chain (shown superimposed on mutant the structure).
In another crystallographic study, we examined the complex between trypsin and the cleaved form of BPTI. Normally, if the cleaved inhibitor binds to trypsin, the enzyme rapidly resynthesizes the peptide bond, but, using a catalytically-inactive trypsin mutant we were able to determine the structure of the complex poised to before the bond is reformed. Together with structures of other structures, from several other laboratories and representing different steps in the mechanism, this structure completes a detailed description of the full catalytic cycle, as shown below.
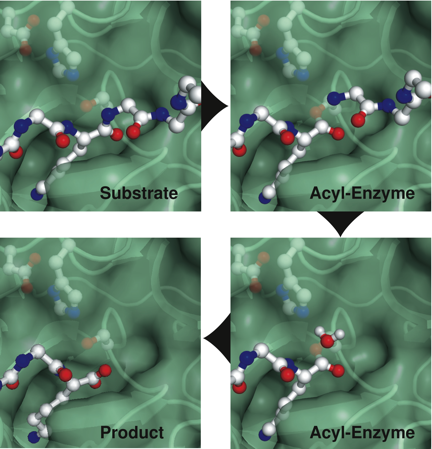
Comparison of these structures demonstrate that this classical catalytic mechanism proceeds with a remarkable economy of molecular motions.
For more information about past and ongoing projects, please see the list of publications.